Eu Mdr Regulation Specific Details : What it is
The Medical Device Regulation (MDR) is a significant regulatory framework that has been implemented by the European Union (EU) to ensure the safety and effectiveness of medical devices. It aims to strengthen and harmonize the regulatory requirements for medical devices across the EU member states. In this article, we will delve into the specific details of the EU MDR regulation, providing you with a comprehensive understanding of what it entails.
1. Overview of the EU MDR

The EU MDR, or European Medical Device Regulation, is a regulation that was adopted in 2017 and came into full effect on May 26, 2021. It replaces the previous Medical Device Directive (MDD) and the Active Implantable Medical Devices Directive (AIMDD). The new regulation has been introduced to address the evolving landscape of medical devices, taking into account technological advancements, market changes, and patient safety concerns.
2. Key Changes and Requirements

The EU MDR brings several key changes and requirements that medical device manufacturers and stakeholders must adhere to. Some of the notable changes include:
- Tighter Classification Rules: The new regulation introduces stricter classification rules based on the potential risks associated with medical devices. It classifies devices into four categories – Class I, Class IIa, Class IIb, and Class III.
- Increased Scrutiny for High-Risk Devices: High-risk devices, such as implantable devices and certain diagnostic products, will undergo a more rigorous assessment process, including a review by expert panels and the European Database on Medical Devices (EUDAMED).
- Enhanced Clinical Evidence Requirements: Manufacturers must provide robust clinical evidence to support the safety and performance of their devices. This includes conducting clinical investigations and post-market surveillance.
- Strengthened Post-Market Surveillance: The EU MDR places a greater emphasis on post-market surveillance activities, such as monitoring the performance and safety of medical devices throughout their lifecycle.
- Unique Device Identification (UDI) System: A comprehensive UDI system will be implemented, enabling the traceability of medical devices and improving their overall transparency and reliability.
3. Impact on Medical Device Manufacturers
The EU MDR has significant implications for medical device manufacturers, both within and outside the EU. Some of the key impacts include:
- Recertification: Manufacturers will need to recertify their existing devices to comply with the new regulations, leading to a considerable financial burden and resource requirement.
- Supply Chain Challenges: The increased scrutiny and documentation requirements may disrupt the supply chain, leading to delays in the availability of medical devices in the EU market.
- Competitive Landscape: Smaller manufacturers may struggle to meet the regulatory requirements, potentially leading to increased consolidation within the industry.
- Resource Allocation: Companies will need to allocate additional resources to ensure compliance with the EU MDR, including hiring regulatory experts and implementing new quality management systems.
FAQs
1. Is compliance with the EU MDR mandatory for all medical device manufacturers?
Yes, compliance with the EU MDR is mandatory for all medical device manufacturers who intend to market their products in the European Union.
2. How long do manufacturers have to transition to the EU MDR?
The transition period for the EU MDR is three years, starting from the date of its full implementation on May 26, 2021. Manufacturers must ensure compliance with the regulation within this timeframe.
3. What are the consequences of non-compliance with the EU MDR?
Non-compliance with the EU MDR can have severe consequences, including the inability to market medical devices within the EU, financial penalties, and damage to the company's reputation.
In conclusion, the EU MDR is a comprehensive regulatory framework that seeks to enhance the safety and effectiveness of medical devices in the European Union. It introduces key changes and requirements that medical device manufacturers must adhere to, ensuring better risk management, robust clinical evidence, and strengthened post-market surveillance. By understanding and complying with the EU MDR, manufacturers can ensure the successful market entry and continued availability of their medical devices in the EU marketplace.
Medical Device Regulation (MDR) | EU-Medizinprodukteverordnung: An- Und
Image Source : www.iww.de Mdr Medical Device Regulation Pdf
 Image Source : why-not.com
Image Source : why-not.com mdr medical device regulation devices pdf vitro diagnostic paper
What Is EU MDR? | Advisera
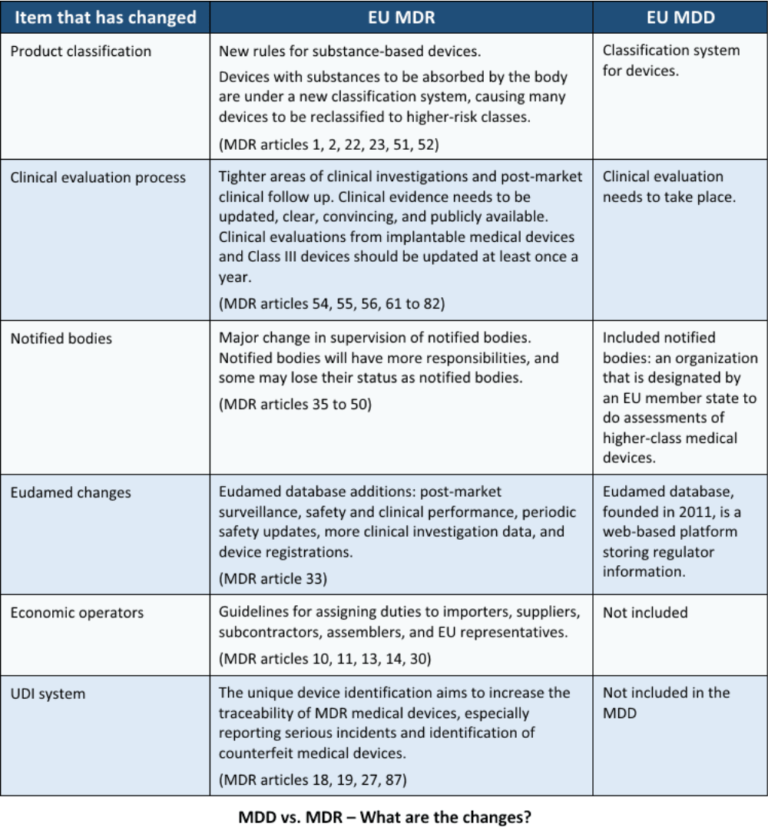 Image Source : advisera.com
Image Source : advisera.com mdr mdd advisera
EU-Medizinprodukteverordnung MDR 2017/745 | DE | TÜV Rheinland
 Image Source : www.tuv.com
Image Source : www.tuv.com Medical Device Regulation Definition | Arena
 Image Source : www.arenasolutions.com
Image Source : www.arenasolutions.com MDR-Zertifikat Für BOWA MEDICAL - BOWA MEDICAL
 Image Source : www.bowa-medical.com
Image Source : www.bowa-medical.com EU MDR – Regulation (EU) 2017/745 | Regulators, Job Board, The European
 Image Source : www.pinterest.com
Image Source : www.pinterest.com MedTech Europe Warns On MDR Implementation Issues | RegDesk
Image Source : www.regdesk.co mdr mdcg warns medtech regdesk ivdr
What is eu mdr?. Mdr medical device regulation devices pdf vitro diagnostic paper. Medical device regulation (mdr). Mdr mdcg warns medtech regdesk ivdr. Mdr medical device regulation pdf