Saudi Arabia Medical Device Regulation
Saudi Arabia Medical Device Regulation: A Comprehensive Guide Introduction: The medical device industry plays a crucial role in the healthcare sector, ensuring the safety and effectiveness of various healthcare treatments. In Saudi Arabia, the Kingdom has implemented robust regulations that govern the sale, import, and distribution of medical devices. These regulations not only prioritize patient safety but also aim to foster innovation and ensure the quality of medical devices available in the Saudi Arabian market. In this post, we will delve into the Saudi Arabia Medical Device Regulation and provide you with a comprehensive understanding of the key aspects, requirements, and procedures involved. Whether you are a healthcare professional, medical device manufacturer, or someone looking to gain insights into the Saudi Arabian medical device market, this guide is here to assist you. 1. Understanding the Regulatory Landscape: In order to successfully navigate the Saudi Arabian medical device market, it is crucial to have a clear understanding of the regulatory landscape. The Saudi Arabia Food & Drug Authority (SFDA) is the regulatory body responsible for overseeing medical device regulations in the Kingdom. SFDA works diligently to ensure that the medical devices available in the market comply with international standards and meet the necessary safety and performance criteria. Key subheading 1: Medical Device Classification Before entering the Saudi Arabian market, medical devices are classified into different categories based on their intended use and potential risks. The classification system includes Classes I, IIa, IIb, III, and IV, with each class having its own set of requirements and regulatory procedures. Here's an overview of the classification criteria: - Class I: Low-risk medical devices such as bandages, non-invasive instruments. - Class IIa: Moderate-risk devices like contact lenses, pregnancy tests. - Class IIb: Devices that are surgically invasive or administer medications, such as pacemakers, surgical instruments. - Class III: High-risk devices that are surgically invasive and have a long-term or permanent effect, such as implantable defibrillators, heart valves. - Class IV: Implantable devices with the highest level of risk, like cochlear implants, artificial hips. 2. Registration and Licensing Process: In order to market and distribute medical devices in Saudi Arabia, manufacturers must obtain the necessary approvals and licenses from SFDA. The registration process involves several steps, including submission of technical documentation, clinical data, and labeling information. Here's an overview of the registration and licensing process: Key subheading 2: Technical Documentation and Clinical Data Submitting comprehensive technical documentation and clinical data is a crucial part of the registration process. This includes detailed information about the design, manufacturing process, performance evaluations, and any relevant testing conducted on the medical device. SFDA requires manufacturers to provide evidence of the device's safety, performance, and effectiveness in order to ensure patient safety. Key subheading 3: Labeling Requirements Accurate and detailed labeling is of utmost importance to ensure the safe use of medical devices. SFDA has specific requirements regarding the information that must be included on the label, such as the device's intended use, instructions for use, warnings, and precautions. It is essential for manufacturers to comply with these labeling requirements to avoid any legal or regulatory issues. 3. Post-Market Surveillance and Vigilance: Once a medical device is approved and enters the market in Saudi Arabia, SFDA maintains strict oversight to ensure the continued safety and effectiveness of the devices. Post-market surveillance involves monitoring the performance, usage, and any reported adverse events related to the medical devices. This allows SFDA to take prompt action in case of any safety concerns or issues. Key subheading 4: Adverse Event Reporting Manufacturers and distributors are required to promptly report any adverse events or unexpected incidents related to their medical devices. SFDA has established a comprehensive adverse event reporting system that allows healthcare professionals and consumers to report any incidents or issues they come across. This reporting system plays a vital role in maintaining the safety and quality of medical devices in Saudi Arabia. Key subheading 5: Product Recalls and Field Corrective Actions In addition to adverse event reporting, SFDA has the authority to issue product recalls or request field corrective actions in cases where there are concerns about the safety or performance of a medical device. Manufacturers must cooperate with SFDA and take immediate action to address the issues raised by the regulatory body. Product recalls and corrective actions are critical in safeguarding patient safety and maintaining the integrity of the medical device market. FAQs: Q1. What is the role of SFDA in regulating medical devices in Saudi Arabia? A1. SFDA is the regulatory body responsible for overseeing medical device regulations in Saudi Arabia. It ensures that medical devices available in the market comply with international standards and meet required safety and performance criteria. Q2. How are medical devices classified in Saudi Arabia? A2. Medical devices in Saudi Arabia are classified into different categories (Class I, IIa, IIb, III, and IV) based on their intended use and potential risks. Each class has its own set of requirements and regulatory procedures. Q3. What are the requirements for medical device registration in Saudi Arabia? A3. The registration process requires manufacturers to submit technical documentation, clinical data, and labeling information to SFDA. This helps ensure the safety, performance, and effectiveness of the medical devices. Q4. What is post-market surveillance and vigilance? A4. Post-market surveillance involves monitoring the performance, usage, and any reported adverse events related to medical devices. SFDA maintains oversight to promptly address any safety concerns or issues. Q5. What should manufacturers do in case of adverse events related to their devices? A5. Manufacturers and distributors are required to promptly report any adverse events or unexpected incidents related to their medical devices. SFDA has an adverse event reporting system in place to ensure timely action and patient safety. Conclusion: The Saudi Arabia Medical Device Regulation plays a crucial role in ensuring patient safety and maintaining the quality of medical devices in the market. By understanding the regulatory landscape, registration process, and post-market surveillance procedures, manufacturers and healthcare professionals can navigate the Saudi Arabian medical device market with confidence. Adhering to these regulations not only helps protect patient health but also contributes to an environment of innovation and excellence in the medical device industry.  Image Source : www.emergobyul.com
Image Source : www.emergobyul.com  Image Source : saudiscoop.com
Image Source : saudiscoop.com 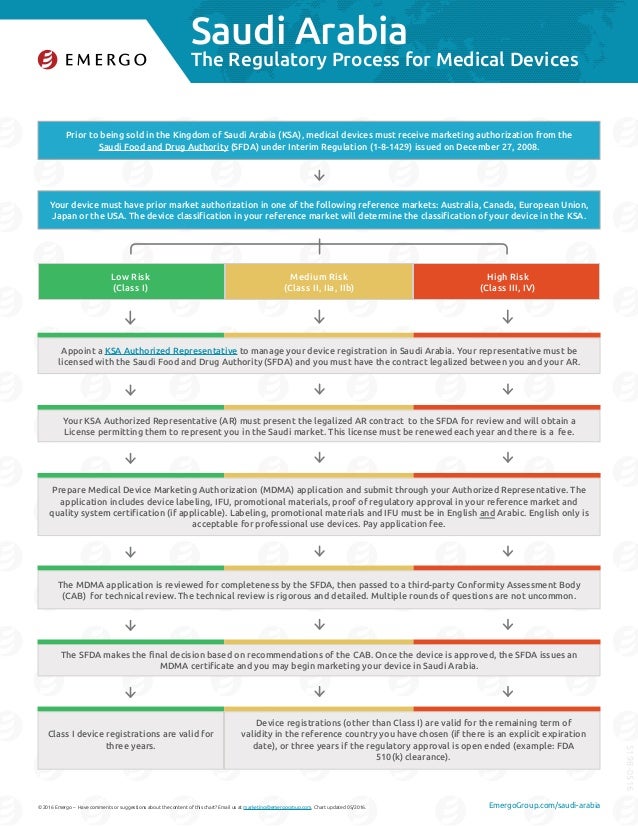 Image Source : www.slideshare.net
Image Source : www.slideshare.net  Image Source : damad.com
Image Source : damad.com  Image Source : operonstrategist.com
Image Source : operonstrategist.com  Image Source : articles.business
Image Source : articles.business  Image Source : www.readkong.com
Image Source : www.readkong.com  Image Source : www.academia.edu
Image Source : www.academia.edu
The Kingdom Of Saudi Arabia (KSA) Has Introduced New Guidance On
Image Source : www.emergobyul.com A Guide To Healthcare Systems In Saudi Arabia | Saudi Scoop
Image Source : saudiscoop.com saudi arabia saudiscoop
Saudi Arabia Medical Device Regulatory Process
Image Source : www.slideshare.net saudi arabia medical device process regulatory emergo slideshare chart
Saudi Arabia Medical Devices Market To Grow At A Significant Rate
Image Source : damad.com Medical Device Registration In Saudi Arabia
Image Source : operonstrategist.com Medical Device Consulting For Riyadh, Saudi Arabia | Operon Strategist
Image Source : articles.business Introduction Of Saudi Arabia Medical Device Regulatory System - Ali Al
Image Source : www.readkong.com (PDF) Saudi Arabia Medical Device Market Outlook 2018 | KuicK Research
Image Source : www.academia.edu The kingdom of saudi arabia (ksa) has introduced new guidance on. Introduction of saudi arabia medical device regulatory system. (pdf) saudi arabia medical device market outlook 2018. Saudi arabia medical device process regulatory emergo slideshare chart. Saudi arabia saudiscoop